- DONNÉES CLINIQUES
4.1 Indications thérapeutiques
DEPO-ELIGARD 22,5 mg est indiqué dans le traitement du cancer de la prostate hormono-dépendant à un stade avancé et dans le traitement du cancer de la prostate hormono-dépendant localisé à risque élevé et localement avancé en combinaison avec la radiothérapie.
4.2 Posologie et mode d’administration
Posologie
Hommes adultes
DEPO-ELIGARD 22,5 mg doit être administré sous la supervision d’un professionnel de la santé qui dispose de l’expérience appropriée pour surveiller la réponse au traitement.
DEPO-ELIGARD 22,5 mg est administré tous les trois mois en injection sous-cutanée unique. La solution injectée forme un dépôt solide de médicament et assure une libération continue d’acétate de leuproréline pendant trois mois.
En règle générale, le traitement du cancer de la prostate avancée par DEPO-ELIGARD 22,5 mg impose un traitement à long terme, lequel ne doit pas être interrompu en cas de rémission ou d’amélioration.
DEPO-ELIGARD 22.5 mg peut être utilisé comme traitement néoadjuvant ou adjuvant en combinaison avec la radiothérapie dans le cancer de la prostate localisé à risque élevé et localement avancé.
La réponse à DEPO-ELIGARD 22,5 mg doit être surveillée en procédant à des examens cliniques et en mesurant les taux sériques de PSA (antigène prostatique spécifique). Les études cliniques ont montré que la testostéronémie augmentait au cours des 3 premiers jours de traitement chez la plupart des patients non orchidectomisés et diminuait ensuite en l’espace de 3 – 4 semaines pour atteindre des valeurs inférieures aux taux de castration médicale. Une fois atteints, ces taux de castration se maintenaient aussi longtemps que le traitement était poursuivi (< 1.0 % d’augmentation de la testostéronémie). Si la réponse d’un patient se révèle sous-optimale, il faut s’assurer que la testostéronémie a atteint des taux de castration ou qu’elle se maintient à ces taux. Étant donné qu’un manque d’efficacité peut résulter d’une préparation, reconstitution ou administration incorrecte du produit, les taux de testostérone doivent être évalués si des erreurs de manipulation sont suspectées ou avérées (voir rubrique 4.4).
Chez des patients atteints d’un cancer de la prostate résistant à la castration métastatique, qui n’ont pas été castrés par chirurgie et qui sont traités par un agoniste de la GnRH, tel que la leuproréline, et qui sont éligibles pour un traitement par inhibiteurs de la biosynthèse des androgènes ou par inhibiteurs des récepteurs des androgènes, le traitement par un agoniste de la GnRH peut être poursuivi.
Population pédiatrique
La sécurité et l’efficacité de DEPO-ELIGARD 22,5 mg chez les enfants âgés de 0 à 18 ans n’ont pas été établies (voir également rubrique 4.3).
Populations spécifiques de patients
Aucune étude clinique n’a été réalisée chez les patients atteints d’insuffisance hépatique ou rénale.
Mode d’administration
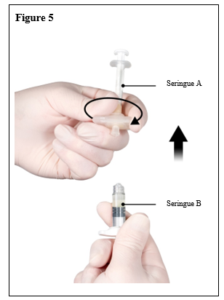
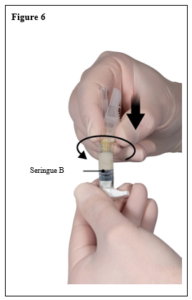
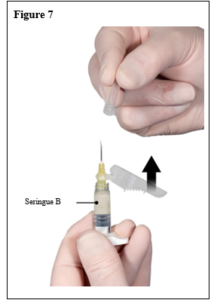
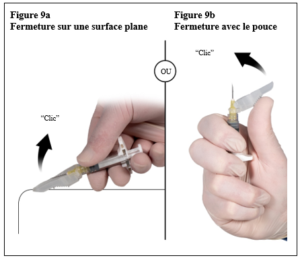
Depo-Eligard 22,5 mg doit être préparé, reconstitué et administré uniquement par des professionnels de la santé habitués à ces procédures.. Les instructions de reconstitution et d’administration doivent être strictement respectées (voir rubrique 4.4 et 6.6). Ne pas administrer le produit s’il n’a pas été préparé correctement.
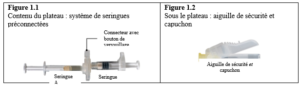
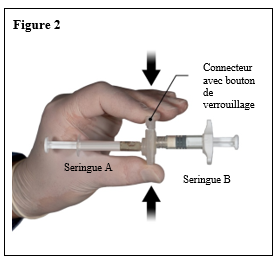
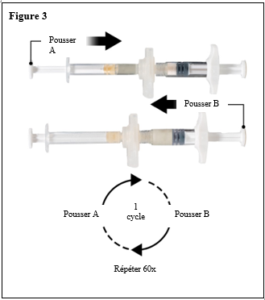
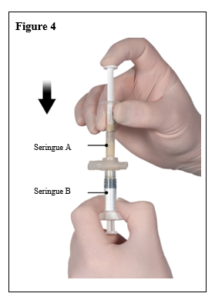
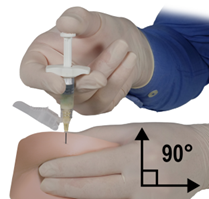
Le contenu de deux seringues stériles préremplies doit être mélangé juste avant l’administration de DEPO-ELIGARD 22,5 mg en injection sous-cutanée.
Au vu des données recueillies dans les expérimentations animales, l’injection intra-artérielle ou intraveineuse doit être absolument évitée.
Comme pour tous les médicaments administrés par voie sous-cutanée, il faut modifier périodiquement le site d’injection.
4.3 Contre-indications
DEPO-ELIGARD 22,5 mg est contre-indiqué chez les femmes et les patients pédiatriques.
Hypersensibilité à l’acétate de leuproréline, aux autres agonistes de la GnRH ou à l’un des excipients mentionnés à la rubrique 6.1.
Patients orchidectomisés (comme avec d’autres agonistes de la GnRH, DEPO-ELIGARD 22,5 mg n’entraîne pas de diminution supplémentaire de la testostéronémie en cas de castration chirurgicale).
Comme traitement unique d’un cancer de la prostate chez les patients souffrant d’une compression médullaire ou de métastases spinales (voir également rubrique 4.4).
4.4 Mises en garde spéciales et précautions d’emploi
Reconstitution correcte :
Des cas d’erreur de manipulation pouvant se produire à n’importe quelle étape du processus de préparation et pouvant conduire à un manque d’efficacité ont été rapportés. Les instructions de reconstitution et d’administration doivent être strictement respectées (voir rubrique 6.6). En cas d’erreur de manipulation suspectée ou avérée, le patient doit faire l’objet d’une surveillance adéquate (voir rubrique 4.2).
Un traitement par suppression androgénique peut allonger l’intervalle QT.
Chez les patients présentant des antécédents ou des facteurs de risques de l’allongement de l’intervalle QT, et chez les patients recevant de manière concomitante des médicaments susceptibles d’allonger l’intervalle QT (voir rubrique 4.5), les médecins doivent évaluer le rapport bénéfice / risque en prenant en compte le risque potentiel de torsades de pointes avant l’initiation du traitement par DEPO-ELIGARD 22,5 mg.
Maladies cardiovasculaires: Un risque accru de développer un infarctus du myocarde, une mort subite d’origine cardiaque et un accident vasculaire cérébral ont été rapportés en association avec l’utilisation d’agonistes de la GnRH chez les hommes. Le risque semble faible sur base des rapports odds ratios, et doit être évalué avec soin avec les facteurs de risque cardiovasculaires pour déterminer un traitement pour les patients atteints de cancer de la prostate. Les patients recevant des agonistes de la GnRH doivent être surveillés pour des signes et symptômes évoquant le développement de maladies cardiovasculaires et être pris en charge conformément à la pratique clinique actuelle.
Poussée transitoire des taux de testostérone : L’acétate de leuproréline, comme les autres agonistes de la GnRH, entraîne une élévation transitoire des concentrations sériques de testostérone, de dihydrotestostérone et des phosphatases acides pendant la première semaine de traitement. Les patients peuvent présenter une aggravation des symptômes ou voir apparaître de nouveaux symptômes tels que douleurs osseuses, neuropathie, hématurie, obstruction urétérale ou vésicale (voir rubrique 4.8). Ces symptômes s’estompent généralement lors de la poursuite du traitement.
L’administration supplémentaire d’un anti-androgène approprié doit être envisagée : commencer 3 jours avant l’instauration du traitement par leuproréline et continuer pendant les deux ou trois premières semaines du traitement. Ce traitement préviendrait les effets de l’élévation initiale de la testostérone sérique.
Après castration chirurgicale, DEPO-ELIGARD 22,5 mg n’entraîne pas de diminution supplémentaire de la testostéronémie chez les sujets masculins.
Densité osseuse : Une diminution de la densité osseuse a été rapportée dans la littérature médicale chez des hommes ayant subi une orchidectomie ou ayant été traités par un agoniste de la GnRH (voir rubrique 4.8).
Le traitement anti-androgène augmente de manière significative le risque de fracture secondaire à une ostéoporose. Il n’existe qu’un nombre limité de données sur la question. Des fractures secondaires à une ostéoporose ont été observées chez 5 % des patients après 22 mois de traitement pharmacologique androgénosuppresseur et chez 4 % des patients après 5 à 10 années de traitement. Le risque de fracture secondaire à une ostéoporose est généralement plus élevé que le risque de fracture pathologique. En dehors d’un déficit prolongé en testostérone, un âge avancé, le tabagisme, la consommation de boissons alcoolisées, l’obésité et le manque d’exercice physique peuvent aussi avoir une influence sur le développement de l’ostéoporose.
Apoplexie hypophysaire: Des cas rares d’apoplexie hypophysaire (un syndrome clinique secondaire à un infarctus de la glande hypophysaire) ont été signalés après l’administration d’agonistes de la GnRH dans le cadre des études de pharmacovigilance. La plupart se sont produits dans les 2 semaines après l’administration de la première dose, quelques-uns dans l’heure qui a suivi. L’apoplexie hypophysaire s’est traduite par des céphalées soudaines, des vomissements, des modifications visuelles, une ophtalmoplégie, une altération de l’état mental et quelquefois un collapsus cardiovasculaire. Ces cas nécessitent une attention médicale immédiate.
Modifications métaboliques: une hyperglycémie et un risque accru de développer un diabète ont été rapportés chez les hommes traités par des agonistes de la GnRH. L’hyperglycémie peut représenter le développement du diabète sucré ou la détérioration du contrôle glycémique chez les patients diabétiques. Surveiller périodiquement la glycémie et / ou l’hémoglobine glyquée (HbA1c) chez les patients traités par un agoniste de la GnRH et gérer avec la pratique courante pour le traitement de l’hyperglycémie ou le diabète. Les modifications métaboliques liées aux agonistes de la GnRH peuvent également inclure la stéatose hépatique.
Convulsions: des rapports post-marketing signalent des crises convulsives chez des patients sous acétate de leuproréline avec ou sans antécédents de facteurs prédisposants. Ces convulsions doivent être prises en charge selon la pratique clinique actuelle.
Hypertension intracrânienne idiopathique: Des cas d’hypertension intracrânienne idiopathique (méningite séreuse) ont été rapportés chez des patients recevant de la leuproréline. Les patients doivent être avertis de la possibilité de signes et symptômes d’hypertension intracrânienne idiopathique, notamment des céphalées sévères ou récurrentes, de troubles visuels et d’acouphènes. En présence d’une hypertension intracrânienne idiopathique, l’interruption du traitement par leuproréline doit être envisagée.
Réactions indésirables cutanées sévères : Des réactions indésirables cutanées sévères (SCAR), dont le syndrome de Stevens-Johnson (SSJ), et la nécrolyse épidermique toxique (NET), qui peuvent engager le pronostic vital ou être fatals, ont été rapportés en association avec le traitement par leuproréline. Au moment de la prescription, les patients doivent être informés des signes et symptômes et surveillés étroitement en cas de réactions cutanées graves. En cas d’apparition de signes et symptômes évocateurs de ces réactions, le traitement par leuproréline doit être arrêté immédiatement et un autre traitement doit être envisagé (le cas échéant).
Autres événements : Des cas d’obstruction urétérale et de compression médullaire, susceptibles d’entraîner une paralysie avec ou sans complications fatales, ont été signalés avec les agonistes de la GnRH. L’apparition d’une compression médullaire ou d’une insuffisance rénale impose l’instauration d’un traitement standard de ces complications.
Les patients qui présentent des métastases vertébrales et/ou cérébrales ou une obstruction des voies urinaires devront être étroitement surveillés pendant les premières semaines de traitement.
4.5 Interactions avec d’autres médicaments et autres formes d’interactions
Aucune étude pharmacocinétique d’interaction médicamenteuse n’a été réalisée avec DEPO-ELIGARD 22,5 mg. Aucune interaction de l’acétate de leuproréline avec d’autres médicaments n’a été signalée.
Le traitement par suppression androgénique étant susceptible d’allonger l’intervalle QT, l’utilisation concomitante de DEPO-ELIGARD 22,5 mg avec des médicaments connus pour allonger l’intervalle QT, ou des médicaments capables d’induire des torsades de pointes tels que les antiarythmiques de classe IA (par exemple quinidine, disopyramide) ou de classe III (par exemple amiodarone, sotalol, dofetilide, ibutilide), la méthadone, la moxifloxacine, les antipsychotiques, etc. doit être évaluée avec précaution (voir rubrique 4.4).
4.6 Fertilité, grossesse et allaitement
Sans objet puisque DEPO-ELIGARD 22,5 mg est contre-indiqué chez les femmes.
4.7 Effets sur l’aptitude à conduire des véhicules et à utiliser des machines
Les effets de DEPO-ELIGARD 22,5 mg sur l’aptitude à conduire des véhicules et à utiliser des machines n’ont pas été étudiés.
Fatigue, vertiges et troubles visuels peuvent entraîner une limitation de l’aptitude à conduire des véhicules et à utiliser des machines. Ces effets peuvent être dus au traitement ou être causés par la maladie sous-jacente.
4.8 Effets indésirables
Les effets indésirables observés avec DEPO-ELIGARD 22,5 mg sont principalement dus à l’action pharmacologique spécifique de l’acétate de leuproréline, à savoir élévations et diminutions de certaines concentrations hormonales. Les effets indésirables les plus fréquemment rapportés sont les bouffées de chaleur, les nausées, malaise, et la fatigue ainsi qu’une irritation locale transitoire au site d’injection. Des bouffées de chaleur légères ou modérées se produisent chez quelque 58 % des patients.
Tableau récapitulatif des effets indésirables
Les événements indésirables suivants ont été rapportés pendant les essais cliniques menés avec DEPO-ELIGARD chez des patients atteints d’un carcinome prostatique avancé. Les événements indésirables sont classés selon les fréquences suivantes : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1,000, < 1/100), rare (≥ 1/10,000, < 1/1,000) et très rare (< 1/10,000), non connu (ne peut être estimé sur la base des données disponibles).
| Tableau 1 : Effets indésirables dans les études cliniques menées avec Depo-Eligard |
| Infections et infestations |
|
| fréquent |
nasopharyngite |
| peu fréquent |
infection urinaire, infection cutanée locale |
| Troubles du métabolisme et de la nutrition |
|
| peu fréquent |
aggravation d’un diabète
|
| Affections psychiatriques |
|
| peu fréquent |
rêves anormaux, dépression, baisse de la libido |
| Affections du système nerveux |
|
|
| peu fréquent |
vertiges, céphalées, insomnie, trouble du goût, trouble de l’odorat, hypoesthésie, sensation de vertiges |
| rare
fréquence indéterminée |
mouvements involontaires anormaux
hypertension intracrânienne idiopathique (méningite séreuse) (voir rubrique 4.4) |
| Affections cardiaques
fréquence indéterminée |
allongement de l’intervalle QT (voir rubrique 4.4 et 4.5) |
| Affections vasculaires |
|
| très fréquent |
bouffées de chaleur |
| peu fréquent |
hypertension, hypotension |
| rare |
syncope, collapsus |
| Affections respiratoires, thoraciques et médiastinales |
|
| peu fréquent
fréquence indéterminée |
rhinorrhée, dyspnée
pneumopathie interstitielle |
| Affections gastro-intestinales |
|
| fréquent |
nausées, diarrhée, gastro-entérite/colite |
| peu fréquent |
constipation, sécheresse buccale, dyspepsie, vomissements |
| rare |
flatulence, éructation |
| Affections de la peau et des tissus sous-cutanés |
|
| très fréquent |
ecchymoses, érythème |
| fréquent |
prurit, sudation nocturne |
| peu fréquent |
moiteur, hyperhidrose |
| rare
fréquence indéterminée |
alopécie, éruption cutanée
syndrome de Stevens-Johnson (SSJ) /nécrolyse épidermique toxique (NET) (voir rubrique 4.4), éruption cutanée toxique, érythème polymorphe |
| Affections musculo-squelettiques et systémiques |
|
| fréquent |
arthralgie, douleur aux membres, myalgie, raideur, faiblesse |
| peu fréquent |
dorsalgie, crampes musculaires |
| Affections du rein et des voies urinaires |
|
| fréquent |
mictions rares, difficultés de miction, dysurie, nycturie, oligurie |
| peu fréquent |
spasmes de la vessie, hématurie, augmentation de la fréquence urinaire, rétention urinaire |
| Affections des organes de reproduction et du sein |
|
| fréquent |
sensibilité mammaire, atrophie testiculaire, douleur dans les testicules,
stérilité, hypertrophie mammaire, dysérection, microcaulie |
| peu fréquent |
gynécomastie, impuissance, troubles testiculaires |
| rare |
douleur mammaire |
| Troubles généraux et anomalies au site d’administration |
|
| très fréquent |
fatigue, brûlure au site d’injection, paresthésie au site d’injection |
| fréquent |
Malaise, douleur au site d’injection, hématome au site d’injection, sensation urticante au site d’injection, |
| peu fréquent |
prurit au site d’injection, induration au site d’injection, léthargie, douleur, pyrexie |
| Rare |
ulcération au site d’injection |
| très rare |
nécrose au site d’injection |
|
Affections hématologiques et du système lymphatique |
|
| fréquent |
modifications hématologiques, anémie
|
| Investigations |
|
| fréquent |
augmentation de la créatinine phosphokinase sanguine, augmentation du temps de coagulation |
| peu fréquent |
augmentation de l’alanine aminotransférase, augmentation des triglycérides sanguins, augmentation du temps de prothrombine, prise de poids |
D’autres événements indésirables qui ont été rapportés en général avec le traitement par l’acétate de leuproréline incluent œdème périphérique, embolie pulmonaire, palpitations, myalgie, faiblesse musculaire, changement dans la sensation de la peau, frissons, éruption cutanée, amnésie et troubles visuels. Une atrophie musculaire a été observée lors de l’utilisation à long terme de produits de cette classe. De rares cas d’apoplexie hypophysaire secondaire à un infarctus ont été rapportés après l’administration d’agonistes de la GnRH de courte et de longue durée d’action. Des cas de thrombocytopénie et de leucopénie ont été rapportés rarement. Des modifications de la tolérance au glucose ont été rapportées.
Des convulsions ont été rapportées après l’administration d’analogues agonistes de la GnRH (voir rubrique 4.4).
Les événements indésirables locaux rapportés après injection de DEPO-ELIGARD 22,5 mg sont semblables à ceux associés aux produits similaires injectés par voie sous-cutanée.
Généralement, ces événements indésirables localisés consécutifs à l’injection sous-cutanée sont légers et décrits comme étant de courte durée.
Dans de rares cas, des réactions anaphylactiques/anaphylactoïdes ont été rapportées après l’administration d’analogues agonistes de la GnRH.
Modifications de la densité osseuse
Une diminution de la densité osseuse a été rapportée dans la littérature médicale chez des hommes qui ont subi une orchidectomie ou qui ont été traités par un analogue de la GnRH. Il est probable qu’un traitement à long terme par leuproréline révèle des signes croissants d’ostéoporose. Pour obtenir plus d’informations sur l’augmentation du risque de fracture secondaire à une ostéoporose, voir rubrique 4.4.
Exacerbation des signes et des symptômes de la maladie
Le traitement par leuproréline peut entraîner une exacerbation des signes et des symptômes de la maladie pendant les premières semaines de traitement. Une aggravation d’affections telles que métastases vertébrales et/ou obstruction urinaire ou hématurie peut faire apparaître des problèmes neurologiques tels que faiblesse et/ou paresthésie des membres inférieurs ou une accentuation des symptômes urinaires.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via :.
Belgique
Agence Fédérale des Médicaments et des Produits de Santé
www.afmps.be
Division Vigilance :
website: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
4.9 Surdosage
DEPO-ELIGARD 22,5 mg ne présente pas de risque d’usage abusif et les surdoses volontaires sont peu probables. Aucun cas d’abus ou de surdose n’a été rapporté dans la pratique clinique avec l’acétate de leuproréline, mais en cas d’exposition excessive, une mise en observation et un traitement de soutien symptomatique sont recommandés.