- KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
DEPO-ELIGARD 22,5 mg is geïndiceerd voor de behandeling van hormoonafhankelijke gevorderde prostaatkanker en voor de behandeling van lokale hormoonafhankelijke prostaatkanker met een hoog risico en lokaal gevorderde hormoonafhankelijke prostaatkanker in combinatie met radiotherapie.
4.2 Dosering en wijze van toediening
Dosering
Volwassen mannen
DEPO-ELIGARD 22,5 mg dient toegediend te worden onder toezicht van een professionele gezondheidszorgverstrekker die over de gepaste ervaring beschikt om de respons op de behandeling te kunnen volgen.
DEPO-ELIGARD 22,5 mg wordt toegediend als een enkele subcutane injectie elke drie maanden. De geïnjecteerde oplossing vormt een vast geneesmiddelendepot en geeft gedurende een periode van drie maanden continu leuprorelineacetaat af.
Als algemene regel houdt een therapie voor gevorderde prostaatkanker met DEPO-ELIGARD 22,5 mg een langetermijnbehandeling in en deze therapie mag niet gestopt worden bij remissie of verbetering.
DEPO-ELIGARD 22,5 mg kan worden gebruikt als neoadjuvante of adjuvante behandeling in combinatie met radiotherapie bij lokale prostaatkanker met een hoog risico en lokaal gevorderde prostaatkanker.
Het antwoord op DEPO-ELIGARD 22,5 mg moet gevolgd worden aan de hand van klinische parameters en door de hoeveelheden van het prostaatspecifieke antigeen (PSA) in het serum te meten. Klinische studies hebben aangetoond dat de testosteronwaarden stegen gedurende de eerste 3 dagen van de behandeling bij de meerderheid van niet-gecastreerde patiënten en dat ze daarna in 3 tot 4 weken daalden tot onder de medische castratiespiegels. Eens bereikt, werden de castratiespiegels behouden zolang de behandeling werd voortgezet (<1.0 % doorbraak van testosteron). Wanneer het antwoord van een patiënt suboptimaal lijkt, zou moeten bevestigd worden dat de serumtestosteronwaarden het castratieniveau bereikt hebben of op dat niveau behouden blijven.
Aangezien een gebrek aan effectiviteit het gevolg kan zijn van onjuiste bereiding, reconstitutie of toediening, moeten in gevallen van vermoede of bekende onjuiste handelingen de testosteronwaarden worden geëvalueerd (zie rubriek 4.4).
Bij patiënten met uitgezaaide castratieresistente prostaatkanker, die niet operatief zijn gecastreerd, en die een GnRH-agonist, zoals leuproreline krijgen, en die in aanmerking komen voor behandeling met androgene biosyntheseremmers of androgene receptorremmers, kan de behandeling met een GnRH- agonist worden voortgezet.
Pediatrische patiënten
De veiligheid en werkzaamheid van DEPO-ELIGARD 22,5 mg bij kinderen van 0 tot 18 jaar zijn niet vastgesteld (zie ook rubriek 4.3)
Specifieke patiëntengroepen
Er werden geen klinische studies uitgevoerd bij patiënten met een gestoorde lever- of nierfunctie.
Wijze van toediening
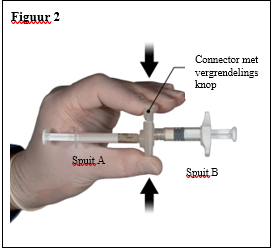
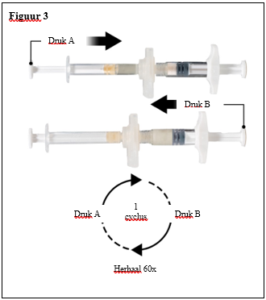
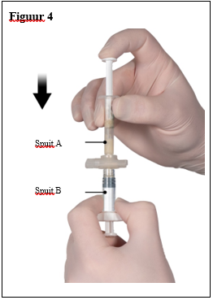
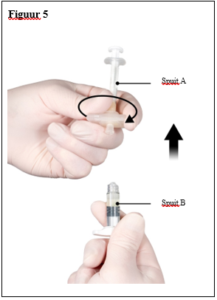
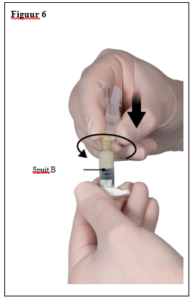
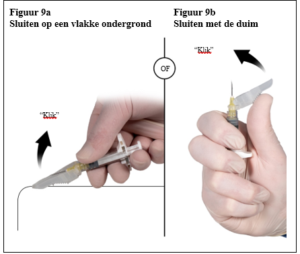
Depo-Eligard 22,5 mg mag uitsluitend worden bereid, gereconstitueerd en toegediend door zorgverleners die bekend zijn met deze procedures. De instructies voor reconstitutie en toediening dienen nauwgezet te worden opgevolgd (zie rubriek 4.4 en 6.6). Als het product niet op de juiste wijze wordt bereid, mag het niet worden toegediend.
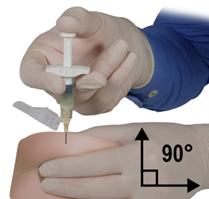
De inhoud van de twee voorgevulde steriele spuiten moet gemengd worden onmiddellijk voor de toediening van DEPO-ELIGARD 22,5 mg door subcutane inject
Intra-arteriële of intraveneuze injecties moeten strikt vermeden worden. Dit is gebleken uit de ervaring met dieren.
Zoals met andere geneesmiddelen die door een subcutane injectie worden toegediend, moet de injectieplaats regelmatig afgewisseld worden.
4.3 Contra-indicaties
DEPO-ELIGARD 22,5 mg is gecontra-indiceerd bij vrouwen en kinderen.
Overgevoeligheid voor leuprorelineacetaat, voor andere GnRH-agonisten of voor (één van) de in rubriek 6.1 vermelde hulpstof(fen).
Bij patiënten die voorafgaand gecastreerd werden (net als andere GnRH-agonisten geeft DEPO-ELIGARD 22,5 mg geen verdere daling van het testosterongehalte in het geval van chirurgische castratie).
Als enige behandeling van prostaatkanker bij patiënten met samendrukking van het ruggenmerg of aangetoonde metastasen in het ruggenmerg (zie ook rubriek 4.4)
4.4 Bijzondere waarschuwingen en voorzorgen bij gebruik
Correcte reconstitutie: Er zijn gevallen gemeld van handelingsfouten, die kunnen optreden bij iedere stap van de bereidingsprocedure en die mogelijk leiden tot gebrek aan effectiviteit. De instructies over reconstitutie en toediening dienen nauwgezet te worden opgevolgd (zie rubriek 6.6). In gevallen van vermoedelijke of bekende handelingsfouten dienen patiënten op gepaste wijze te worden gemonitord (zie rubriek 4.2).
Androgeendeprivatietherapie kan het QT-interval verlengen.
Bij patiënten met een voorgeschiedenis van of risico op QT verlenging en bij patiënten die gelijktijdig geneesmiddelen gebruiken die mogelijk het QT-interval kunnen verlengen (zie rubriek 4.5), dient de arts de baten/risicoverhouding, inclusief de kans op torsade de pointes te evalueren voorafgaand aan de start van DEPO-ELIGARD 22,5 mg.
Cardiovasculaire aandoeningen: er is een verhoogd risico op het ontwikkelen van een myocardinfarct, plotse hartdood en beroerte geassocieerd met het gebruik van GnRH-agonisten bij mannen gemeld. Het risico lijkt gering op basis van de gerapporteerde odds ratio’s, en dient zorgvuldig te worden geëvalueerd samen met de cardiovasculaire risicofactoren bij het bepalen van een behandeling voor patiënten met prostaatkanker. Patiënten die behandeld worden met GnRH-agonisten dienen gecontroleerd te worden op symptomen en tekenen die wijzen op de ontwikkeling van cardiovasculaire aandoeningen en behandeld te worden volgens de huidige klinische praktijk
Transiënte verhoging van testosteron: Leuprorelineacetaat veroorzaakt, net als andere GnRH-agonisten, een tijdelijke verhoging van de serumconcentraties van testosteron, dihydrotestosteron en zuur fosfatase tijdens de eerste behandelingsweek. Patiënten kunnen ervaren dat de symptomen verslechteren of dat er nieuwe verschijnen, waaronder botpijn, neuropathie, hematurie of ureter- of blaasobstructie (zie rubriek 4.8). Deze symptomen verdwijnen gewoonlijk bij voortzetting van de behandeling.
Bijkomende toediening van een gepast antiandrogeen moet overwogen worden: te beginnen 3 dagen voor de start van de leuprorelinebehandeling en verder te geven gedurende de eerste twee tot drie weken van die behandeling. Dit werd gerapporteerd om de gevolgen van de initiële stijging van serumtestosteron tegen te gaan.
Na chirurgische castratie leidt DEPO-ELIGARD 22,5 mg niet tot een verdere daling van de serumtestosteronwaarden bij mannelijke patiënten.
Botdensiteit: Verminderde botdensiteit bij mannen werd gerapporteerd in de medische literatuur na een castratie of na behandeling met GnRH-agonisten (zie rubriek 4.8).
Antiandrogeentherapie verhoogt significant het risico op breuken door osteoporose. Er zijn hierover slechts beperkte gegevens beschikbaar. Breuken door osteoporose werden vastgesteld bij 5% van de patiënten na 22 maanden van farmacologische androgeendeprivatietherapie en bij 4% van de patiënten na 5 tot 10 jaar behandeling. Het risico op breuken door osteoporose is over het algemeen hoger dan het risico op pathologische breuken. Naast een langdurig tekort aan testosteron kunnen verhoogde leeftijd, roken en alcoholinname, obesitas en onvoldoende lichaamsbeweging een invloed hebben op de ontwikkeling van osteoporose.
Hypofyse apoplexie: Tijdens postmarketingonderzoek werden zeldzame gevallen van hypofysaire apoplexie (een klinisch syndroom voortkomend uit een bloeding van de hypofyse) gemeld na de toediening van GnRH-agonisten. Het merendeel van de gevallen trad op binnen 2 weken na de eerste dosis en enkele gevallen deden zich binnen het eerste uur voor. In deze gevallen werd hypofysaire apoplexie gekenmerkt door plotselinge hoofdpijn, braken, visuele veranderingen, oftalmoplegie, verandering in mentale toestand en soms cardiovasculaire collaps. Directe medische hulp is vereist.
Metabole veranderingen: hyperglycemie en een verhoogd risico op het ontwikkelen van diabetes zijn gemeld bij mannen die behandeld worden met GnRH-agonisten. Hyperglycemie kan duiden op de ontwikkeling van diabetes mellitus of een verslechtering van de glycemische controle van patiënten met diabetes. Controleer periodiek de bloedglucose en/of het gehalte geglycosyleerd hemoglobine (HbA1c) bij patiënten die een GnRH agonist toegediend krijgen en behandel volgens de huidige klinische praktijk de hyperglycemie of diabetes. Metabole veranderingen in verband met GnRH-analogen kunnen ook leververvetting omvatten.
Convulsies: Tijdens postmarketingonderzoek werden convulsies waargenomen bij patiënten onder leuprorelineacetaat met of zonder voorgeschiedenis van predisponerende factoren. Deze convulsies moeten worden beheerst volgens de huidige klinische praktijken.
Idiopathische intracraniële hypertensie: Idiopathische intracraniële hypertensie (pseudotumor cerebri) is gemeld door patiënten die leuproreline ontvingen. Patiënten moeten worden gewaarschuwd voor verschijnselen en symptomen van idiopathische intracraniële hypertensie, waaronder ernstige of terugkerende hoofdpijn, zichtstoornissen en tinnitus. Indien idiopathische intracraniële hypertensie optreedt, moet worden overwogen het gebruik van leuproreline te staken.
Ernstige huidreacties: In verband met de behandeling met leuproreline zijn ernstige huidreacties (SCARs) gemeld, waaronder Stevens-Johnson-syndroom (SJS) en toxische epidermale necrolyse (TEN), die levensbedreigend of fataal kunnen zijn. Wanneer dit geneesmiddel wordt voorgeschreven,
dienen patiënten te worden geadviseerd over de tekenen en symptomen. Zij moeten nauwlettend worden gecontroleerd op huidreacties. Als er tekenen en symptomen optreden die op deze reacties wijzen, dan moet leuproreline onmiddellijk worden stopgezet en een alternatieve behandeling worden overwogen (zoals geëigend).
Andere voorvallen: Gevallen van ureterobstructie en samendrukking van het ruggenmerg, die kunnen bijdragen tot verlamming, al dan niet fataal, werden gerapporteerd met GnRH-agonisten.
Wanneer zich een samendrukking van het ruggenmerg of nierfalen ontwikkelt, moet een standaardbehandeling voor deze complicaties worden opgestart.
Patiënten met vertebrale en/of hersenmetastasen, evenals patiënten met urinewegobstructie, moeten van nabij gevolgd worden gedurende de eerste behandelingsweken.
4.5 Interacties met andere geneesmiddelen en andere vormen van interactie
Er werden geen farmacokinetische geneesmiddel-geneesmiddel interactiestudies uitgevoerd met DEPO-ELIGARD 22,5 mg. Er zijn geen rapporten beschikbaar over enige interactie van leuprorelineacetaat met andere geneesmiddelen.
Omdat een androgeendeprivatietherapie het QT-interval kan verlengen, dient het gelijktijdig gebruik van Depo-Eligard 22,5 mg en geneesmiddelen waarvan bekend is dat zij het QT-interval verlengen of geneesmiddelen die torsade de pointes kunnen induceren zoals Klasse IA-geneesmiddelen (bijv. kinidine, disopyramide) of Klasse III-geneesmiddelen (bijv. amiodaron, sotalol, dofetilide, ibutilide), anti-aritmische geneesmiddelen, methadon, moxifloxacine, anti-psychotica, etc. zorgvuldig afgewogen te worden (zie rubriek 4.4).
4.6 Vruchtbaarheid, zwangerschap en borstvoeding
Niet van toepassing want DEPO-ELIGARD 22,5 mg is gecontra-indiceerd bij vrouwen.
4.7 Beïnvloeding van de rijvaardigheid en het vermogen om machines te bedienen
Er werden geen studies uitgevoerd om het effect van DEPO-ELIGARD 22,5 mg op de rijvaardigheid en het vermogen om machines te bedienen te bestuderen.
De rijvaardigheid en het gebruik van machines kunnen bemoeilijkt worden door vermoeidheid, duizeligheid en stoornissen in het zicht. Dit kunnen bijwerkingen zijn van de behandeling, maar kan ook te wijten zijn aan de onderliggende ziekte.
4.8 Bijwerkingen
Bijwerkingen die werden gezien met DEPO-ELIGARD zijn voornamelijk toe te schrijven aan de specifieke farmacologische werking van leuprorelineacetaat, met name stijging en daling van bepaalde hormoonconcentraties. De meest beschreven bijwerkingen zijn warmteopwellingen, misselijkheid, een gevoel van onbehagen en vermoeidheid en voorbijgaande lokale irritatie op de injectieplaats. Milde of gematigde warmteopwellingen komen voor bij ongeveer 58% van de patiënten.
Overzicht van bijwerkingen in tabelvorm
De volgende bijwerkingen werden gerapporteerd tijdens klinische studies met DEPO-ELIGARD bij patiënten met gevorderde prostaatkanker. Bijwerkingen worden geklasseerd volgens frequentie als volgt: zeer vaak (≥1/10), vaak (≥1/100, <1/10), soms (≥1/1000, <1/100), zelden (≥1/10.000, <1/1000) en zeer zelden (<1/10.000) of niet gekend (kan niet geschat worden op basis van beschikbare gegevens).
| Tabel 1: Bijwerkingen in klinische studies met Depo-Eligard |
| Infecties en parasitaire aandoeningen |
|
| vaak |
nasofaryngitis |
| soms |
urineweginfectie, lokale huidinfectie |
| Voedings- en stofwisselingsstoornissen |
|
| soms |
verergering van diabetes mellitus |
| Psychische stoornissen |
|
| soms |
abnormale dromen, depressie, verminderd libido |
| Zenuwstelselaandoeningen |
|
|
| soms |
duizeligheid, hoofdpijn, slapeloosheid, verstoring van smaak- en reukzin, hypoaesthesie, vertigo |
| zelden
niet bekend |
abnormale onwillekeurige bewegingen
idiopathische intracraniële hypertensie (pseudotumor cerebri) (zie rubriek 4.4) |
| Hartaandoeningen
niet bekend |
QT verlenging (zie rubriek 4.4 en 4.5) |
| Bloedvataandoeningen |
|
| zeer vaak |
warmteopwellingen |
| soms |
hypertensie, hypotensie |
| zelden |
syncope, collaps |
| Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen |
|
| soms
niet bekend |
rinorroe, dyspneu
interstitiële longziekte |
| Maagdarmstelselaandoeningen |
|
| vaak |
misselijkheid, diarree, gastro-enteritis/colitis |
| soms |
constipatie, droge mond, dyspepsie, braken |
| zelden |
flatulentie, oprispingen |
| Huid- en onderhuidaandoeningen |
|
| zeer vaak |
ecchymoses, erytheem |
| vaak |
pruritus, nachtelijk zweten |
| soms |
klamheid, verhoogd zweten |
| zelden
niet bekend |
alopecie, huideruptie
Stevens-Johnson syndroom / toxische epidermale necrolyse (SJS/TEN) (zie rubriek 4.4), toxische huideruptie, erythema multiforme |
| Skeletspierstelsel- en bindweefselaandoeningen |
|
| vaak |
arthralgie, pijn in de ledematen, myalgie, stijfheid, slapheid |
| soms |
rugpijn, spierkrampen |
| Nier- en urinewegaandoeningen |
|
| vaak |
weinig plassen, moeilijke urinelozing, dysurie, nocturie, verminderde urineproductie |
| soms |
blaaskrampen, bloedplassen, verhoogde plasfrequentie, urineretentie |
| Voortplantingsstelsel- en borstaandoeningen |
|
| vaak |
gevoelige borsten, testiculaire atrofie, pijn in de teelballen, onvruchtbaarheid, hypertrofie van de borsten, erectiele dysfunctie, verminderde penisgrootte |
| soms |
gynaecomastie, impotentie, testiculaire aandoeningen |
| zelden |
pijnlijke borsten |
| Algemene aandoeningen en toedieningsplaatsstoornissen |
|
| zeer vaak |
vermoeidheid, branderig gevoel op de plaats van de injectie, paresthesie op de plaats van de injectie |
| vaak |
malaise, pijn op de plaats van injectie, blauwe plekken op de plaats van injectie, een stekend gevoel op de plaats van injectie |
| soms |
jeuk op de plaats van de injectie, injectieplaatsverharding, lethargie, pijn, koorts |
| zelden |
zweren op de plaats van de injectie |
| zeer zelden |
necrose op de plaats van de injectie |
| Bloed- en lymfestelselaandoeningen |
|
| vaak |
veranderingen in het bloedbeeld, anemie |
| Onderzoeken |
|
| vaak |
verhoogde creatinine fosfokinase bloedspiegels, verlengde bloedstollingstijd |
| soms |
verhoogd alanine aminotransferase, verhoogd bloedtriglyceridengehalte, verlengde prothrombinetijd, gewichtstoename |
Andere bijwerkingen die algemeen vastgesteld werden tijdens een behandeling met leuprorelineacetaat omvatten perifeer oedeem, pulmonaire embolie, palpitaties, myalgie, spierzwakte, wijziging in de huidsensatie, rillingen, uitslag, amnesie en gezichtsstoornissen. Spieratrofie werd waargenomen bij langdurig gebruik van de producten van deze klasse. Een infarct van reeds voorafbestaande hypofysaire adenomen werd zelden gerapporteerd na toediening van zowel lang- als kortwerkende GnRH-agonisten. Er zijn zeldzame rapporten van trombocytopenie en leukopenie. Veranderingen in glucosetolerantie werden gerapporteerd.
Convulsies werden gemeld na toediening van GnRH-agonistanalogen (zie rubriek 4.4).
Lokale bijwerkingen die werden beschreven na de injectie van DEPO-ELIGARD 22,5 mg zijn vergelijkbaar met deze die frequent geassocieerd worden met gelijkaardige subcutaan te injecteren preparaten.
Algemeen zijn deze lokale bijwerkingen na subcutane injectie mild en beschreven als zijnde van korte duur.
Anafylactische/anafylactoïde reacties werden zelden gemeld na toediening van GnRH-agonistanalogen.
Veranderingen in botdensiteit
Verminderde botdensiteit bij mannen wordt gerapporteerd in de medische literatuur na castratie of na behandeling met een GnRH-analoog. Het kan verwacht worden dat een langetermijnbehandeling met leuproreline een stijging van de tekens van osteoporose kan veroorzaken. Wat het verhoogde risico op breuken door osteoporose betreft, zie rubriek 4.4.
Opflakkering van de tekenen en symptomen van de ziekte
Een behandeling met leuproreline kan opflakkeringen van de tekenen en symptomen van de ziekte veroorzaken gedurende de eerste behandelingsweken. Wanneer vertebrale metastasen en/of urinewegobstructie of hematurie verergeren, kunnen neurologische problemen optreden zoals zwakte en/of paresthesie van de onderste ledematen of verslechtering van de urinaire symptomen.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via:
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
4.9 Overdosering
DEPO-ELIGARD 22,5 mg leent zich niet voor misbruik en doelbewuste overdosis is onwaarschijnlijk. Er zijn in de klinische praktijk geen meldingen van misbruik of overdosis bij gebruik van leuproreline-acetaat. Indien er echter toch sprake is van overmatig gebruik, worden observatie en symptoombehandeling aanbevolen.